
Active Projects
Research overview. We work in the area of computational biology to accelerate scientific discovery by developing new scalable and rigorous algorithms. Forty years ago, it would have taken two years to determine just 24 characters of a DNA molecule. Fast forward to today, a single instrument produces more than a trillion characters per day. Genome sequencing, which used to be a centralised process, is now ubiquitous. For instance, the GenomeIndia project alone, generated petabytes of sequencing data. Interpreting DNA molecules from the raw sequencing data is key to understanding genetic mutations associated with diseases such as cancer.
As a result, scalable algorithms and software are indispensable components of computational biology. Our research within this area builds on and extends approximate similarity search, string, geometric, combinatorial, and graph algorithms. Through interdisciplinary collaborations, we have developed new algorithms and widely-used bioinformatics software to improve the accuracy and speed of sequence analysis. Our active projects and their corresponding papers from our lab are listed below.
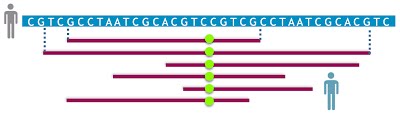
Algorithmic techniques for genome resequencing. Mapping sequences to a reference genome is often the first computational step in deriving biological insights from genomic data. Accurate mapping of sequences is key to predict genetic or epigenetic variation. Doing this precisely has been challenging as human and other mammalian genomes are riddled with near-identical repetitive sequences. Such repeats often confuse existing sequence mappers, resulting in false-positive alignments and poor confidence scores. Desirable characteristics of a mapper include: (i) efficiency of the algorithm, (ii) high sensitivity and specificity, and (iii) scalability to large data sets and human genomes. In this project, we aim to develop provably-good and practical ‘repeat-aware’ mapping algorithms that meet these expectations.
- Weighted minimizer sampling improves long read mapping ISMB 2020
- Strain-level metagenomic assignment and compositional estimation for long reads Nature Comm. 2019
- A fast approximate algorithm for mapping long reads to large reference databases RECOMB 2017
Algorithms and data structures for population genomics. We expect availability of >100 million human genomes in near future. Until recently, algorithms for mapping reads have assumed a single reference genome. To account for the extensive genomic variation present across multiple individuals, read mapping and genotyping efforts must shift to a collection of reference genomes compactly represented using a pangenome graph data structure. This transition requires urgent development of new formats, data structures, algorithms and well-engineered implementations. This project aims to address several related open problems.
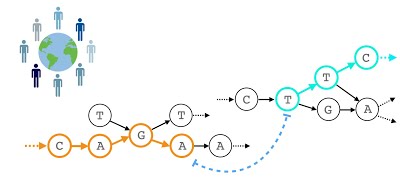
These include (i) development of theoretically well-founded sequence mapping algorithms and heuristics for genome-graphs, (ii) quantification of the benefit of adopting pan-genome approaches, and (iii) designing algorithms to build pan-genome graphs using millions of genomes as input. In each of the three aims, synergy is desired with downstream biological and clinical applications.
- Integer programming framework for pangenome-based genome inference RECOMB 2025
- Haplotype-aware sequence alignment to pangenome graphs RECOMB 2024
- Gap-Sensitive Colinear Chaining Algorithms for Acyclic Pangenome Graphs RECOMB 2023
Approximate techniques to scale genome-to-genome comparisons. Bacterial and viral genomic databases continue to grow exponentially as microbiologists are increasingly turning to whole-genome sequencing driven approaches to address fundamental questions associated with ecology and diversity. Whether it is a disease outbreak or a metagenomics survey of agriculture soil, these investigations typically involve comparing newly sequenced DNA samples against a catalogue of all available annotated genomes. In this project, we seek to develop fast approximation algorithms that enable rapid genome-to-genome comparisons. In other domains such as world-wide-web, approximation algorithms such as locality sensitivity hashing have been key to scale nearest-neighbour document search for web queries.

However, unlike web documents, DNA sequences do not follow a similar structure, e.g., they are not split into tokens or words. We are exploring ways to adapt these techniques for genome sequences while addressing domain-specific challenges.
- High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries Nature Comm. 2018
- A fast adaptive algorithm for computing whole-genome homology maps ECCB 2018
Parallel computing to accelerate bioinformatics. In recent years, portable and real-time DNA/RNA sequencing devices have become a reality. For time-critical applications in healthcare, it is important to achieve real-time computing ability to offer point-of-care diagnostics. Given the ongoing trends in hardware design of CPUs and GPUs, a sequential algorithm can achieve at most a tiny fraction of the available performance. The goal of this project to resolve computational bottlenecks in genomics by designing parallel architecture-aware algorithms.
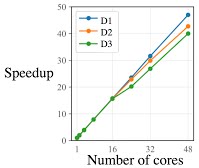
As such, this project entails inventing new parallel algorithms with guarantees of work-optimality (whenever possible) for a variety of string and graph-based methods that are prevalent in bioinformatics. In the near term, we are looking at accelerating long-read mapping and genome assembly algorithms, both of which require significant computing time and resources.
- Accelerating minimap2 for long-read sequencing applications on modern CPUs Nature Comp. Sci. 2022
- Accelerating sequence alignment to graphs IPDPS 2019
- A parallel connectivity algorithm for de Bruijn graphs in metagenomic applications ACM SC 2015
- Fine-grained GPU parallelization of pairwise local sequence alignment IEEE HiPC 2014
Collaborators
Multidisciplinary cooperation is critical in bioinformatics. We enjoy working with scientists bringing complementary expertise from both industry and academia.
- Parallel Computing Lab, Intel, India
- Strand Life Sciences, India
- Environmental Microbial Genomics Lab, Georgia Tech, USA
- Laboratory of AI in Genomics, Genome Institute of Singapore
- University Hospital of Dusseldorf, Germany